
30 Jul Research: Project SCN1A-UP! Therapeutic strategies for Dravet syndrome
The project SCN1A-UP!, coordinated by Dr Massimo Mantegazza and funded by the EJPRD 2020, is a collaborative research involving six European research groups and five Patients Advocacy Organizations.
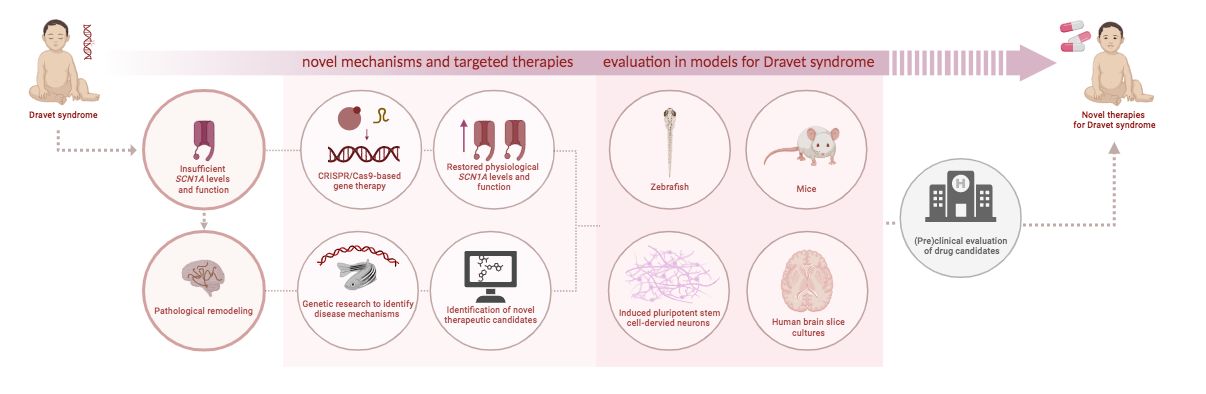
The overall objective of SCN1A-UP! has been to perform pre-clinical studies for developing more effective treatments for Dravet Syndrome with a bottom up approach, targeting SCN1A loss of function, the primary cause of the disease, and pathologic remodeling that can develop at later stages, therapeutic concepts that could be applied also to other diseases.
The results of the project will be presented and discussed during the final meeting, which will be held in Rome, Italy, on 10 September. Here you can find the meeting program
Below is an infographic summarizing the study objectives and design and a wider description of them, already anticipated in brief at the start of the project.
The Consortium
The European Consortium SCN1A-UP! has been funded by the European Joint Programme on Rare Diseases (EJPRD) and consists of 6 European research groups:
Consortium Partners
- Massimo Mantegazza (Coordinator), Centre national de la recherche scientifique (CNRS) [France]
- Vania Broccoli, Ospedale San Raffaele [Italy]
- Peter de Witte, University of Leuven [Belgium]
- Nael Nadif Kasri, Radboud University [The Netherlands]
- Ulrike Hedrich-Klimosch, University of Tübingen [Germany]
- Eleonora Aronica, University of Amsterdam [The Netherlands]
Patients Advocacy Organisations Involved
- Gruppo Famiglie Dravet APS [Italy]
- Alliance Syndrome de Dravet (ASD) [France]
- Associazione Italiana sindrome di Dravet [Italy]
- Dravet-Syndrom e.V. [Germany]
- Stichting Dravetsyndroom [The Netherlands]
Project objectives
The Consortium objective is to develop innovative therapies for Dravet syndrome.
Particularly, the objective of the SCN1A-UP! Consortium is to discover effective disease-preventing or modifying treatments performing pre-clinical studies. Clinical assessments in humans, which is outside the scope of this consortium, will be necessary in order to ultimately make these groundbreaking disease-modifying therapies available to patients in need.
Genetic diseases cause initial dysfunctions that are directly induced by the pathogenic genetic variant (mutation). In the case of Dravet syndrome, pathogenic variants of the SCN1A gene induce loss of function of the NaV1.1 sodium channel leading to hypoexcitability (reduced activity) of GABAergic neurons. The function of GABAergic neurons is to reduce and/or regulate the excitability of neuronal networks. They are present in all brain areas.
The initial dysfunctions directly induced by the genetic mutation can generate different types of secondary responses (remodeling):
a) some that amplify pathological1 modifications due to the gene variants, thus worsening the symptoms of the disease (pro-pathologic remodeling)
b) some that tend to counteract those pathological modifications, thus ameliorating the symptoms of the disease (homeostatic2 remodeling).
The research group hypothesizes that these secondary changes are due to the brain’s (mal)adaptive responses to the expression3 of the SCN1A variants, and these secondary changes can contribute to the clinical features of Dravet syndrome (severity of symptoms).
According with this hypothesis, in a poly-therapeutic approach, it might benefit patients to treat not only the malfunctioning of the SCN1A gene but also the secondary responses with the aim of inhibiting pro-pathological responses (a) and amplifying homeostatic responses (b).
1Abnormal anatomical or physiological conditions that lead to a disease.
2Secondary response that tends to re-establish the normal condition of an organ, so that to maintain a state of balance among all the body systems needed for the body to survive and function correctly
3Gene expression is the process by which the information encoded in a gene is turned into a function (NaV1.1 sodium channel protein in the case of SCN1A gene). This mostly occurs via the transcription of RNA molecules that code for proteins (or non-coding RNA molecules that serve other functions).
Study design
SCN1A-UP! is targeting all these aspects. To achieve this, the research group is currently assessing two distinct therapeutic strategies:
1) to reduce initial dysfunctions of the SCN1A gene
2) to reduce pro-pathologic remodeling and to enhance homeostatic remodeling, both caused by secondary responses.
1) to reduce initial dysfunctions of the SCN1A gene
This strategy aims to directly address the initial cause of the disease, which is the loss-of-function of SCN1A/NaV1.1 (causing insufficient production of the NaV1.1 protein). Using gene-therapy methods, the research group selectively and physically targets the SCN1A gene to increase its natural transcription, in order to augment gene expression and ultimately the amount of the NaV1.1 protein produced by the cells. Thus, the objective here is to overcome SCN1A/NaV1.1 loss-of-function by restoring the normal levels of functional NaV1.1, and the research group is developing two specific molecular tools that can be delivered with viral vectors (gene therapy).
The first molecular tool exploits the CRISPR-Cas9 system, using a modified version (known as CRISPR-ON or activatory CRISPR, CRISPRa) that allows control of gene expression with high specificity by binding to the regulatory areas (promoters) of the SCN1A gene, increasing transcription, with the goal to increase the NaV1.1 protein. The method is based on the delivery with viral vectors of genetic material that leads to the expression in the cells of modified bacterial proteins that can enhance gene transcription and of an RNA molecule (denominated gRNA) that allows the specific interaction of these proteins with the SCN1A regulatory areas. The researchers have tested the method in cultures of mouse neocortical neurons and of induced pluripotent stem cell-derived human neurons, observing robust increase of SCN1A transcription and NaV1.1levels. However, the efficiency in vivo in mouse models is low because the system is large and it is difficult to pack it into the small viral vectors (AAV) currently used for gene therapy. The research group is optimizing the system for reducing its size.
In parallel, they are developing a second molecular tool using small domains of mammalian proteins: a domain called Zinc-finger that selectively binds to the regulatory areas of SCN1A, fused to a protein domain called VP64, which is an activator of the transcription of the gene. This molecular tool is much smaller than CRISPR-ON (the first molecular tool introduced above) and it can be directly inserted into AAV viral vectors to be administered to patients. The research group has already validated it in cell cultures (as above), observing a very good increase of SCN1A transcription and NaV1.1levels. They are now evaluating the effect in vivo in mouse models and in the other experimental models (animal and cell models of Dravet syndrome) of the SCN1A-UP! consortium.
4Transcription is the process of making an RNA copy of a gene’s DNA sequence. This copy, called messenger RNA (mRNA), carries the gene’s protein information encoded in DNA. The purpose of transcription is to produce an mRNA copy of a gene, to allow the genetic information to pass out of the nucleus, through the nuclear pores where it can be used to assemble a protein.
5The CRISPR/Cas-9 system consists of two elements: the guide RNA (gRNA) used to locate (bind) the target DNA to which to address the second element, the Cas-9 protein can either cut or modify the DNA at the location identified by guide RNA or interact with other endogenous elements that can modulate (e.g. increase) the expression of a gene.
2) to reduce pro-pathologic remodeling and to enhance homeostatic remodeling, both caused by secondary responses.
The second strategy involves the identification of the secondary responses, thus the novel modifications (pro-pathologic and homeostatic remodeling) arising secondarily to the causative SCN1A genetic variant and amenable to therapeutic intervention.
As part of this strategy, the consortium is pursuing two different approaches.
The first approach within this strategy investigates the remodeling of other genes’ expression levels. In particular, through the analysis of genetic information obtained from a zebrafish model of Dravet syndrome, the research group has identified dysregulated genes (with impaired functioning) implicated in remodeling resulting from SCN1A loss-of-function. These discoveries are used as input for computational analyses identifying novel therapeutic candidates, including repurposed drugs (use of existing drugs for new therapeutic purposes, which can have fast translational potential and made available to patients more rapidly) that can counteract identified pro-pathologic remodeling.
They also aim to compare the remodeling of gene expression observed in the different experimental models of the consortium, including in mouse models where they have already identified remodeling candidates for therapeutic intervention. The research group has tested some candidate drugs in zebrafish, which they are now validating on mouse models.
In a second parallel approach, they have performed functional studies (evaluation of the properties of the neurons in mouse models) for disclosing secondary responses. They have identified a homeostatic response that they are aiming to boost in a therapeutic approach (the research group already obtained promising results in chronic treatments of mice: reduction of seizures and mortality, amelioration of behavioral/cognitive deficits without signs of toxicity).
All the treatment strategies identified in the project are currently being evaluated using various animal and cell models of Dravet syndrome, including zebrafish and mouse models, induced pluripotent stem cell-derived human neurons, as well as human brain slice cultures. The broad spectrum of preclinical models within a consolidated pipeline is a strength of the SCN1A-UP! project. Thorough validation of candidate therapies, including lead efficacy and dosage optimization, will be carried out exploiting this pipeline.